The cytoplasm surrounds the nucleus, and it is in the
cytoplasm that the work of the cell takes place. Embedded
in the cytoplasm are various membrane-enclosed
compartments (endoplasmic reticulum, Golgi apparatus,
mitochondria, lysosomes) that function as the organs of
the cells. Most organelles are surrounded by one or two lipid membranes, similar to plasma membrane, that separate
the organelles from the cytosol.
Ribosomes, Endoplasmic Reticulum,
and Golgi Apparatus
The ribosomes, endoplasmic reticulum, and Golgi apparatus
represent the primary sites of protein synthesis in
the cell (Fig. 1-4). Because these organelles lack direct
communication with the cytosol, they use transport vesicles
to move newly synthesized proteins, membrane components,
and soluble molecules from one organelle to
another. These transport vesicles bud off from the membrane
of one organelle and fuse with another, carrying
the transported material.
Ribosomes(definition and it functions): The ribosomes are small particles of nucleoproteins
(rRNA and proteins) that are held together by
a strand of mRNA to form polyribosomes (also called
polysomes). Polysomes exist as isolated clusters of free
ribosomes within the cytoplasm or attached to the membrane
of the endoplasmic reticulum (Fig. 1-4). Free ribosomes
are involved in the synthesis of proteins, mainly
enzymes that aid in the control of cell function, whereas
those attached to the endoplasmic reticulum translate
mRNAs that code for proteins secreted from the cell
or stored within the cell (e.g., granules in white blood
cells).
Endoplasmic Reticulum(definition and it functions) : The endoplasmic reticulum
(ER) is an extensive system of paired membranes and
flat vesicles that connect various parts of the inner cell (Fig. 1-4). Between the paired ER membranes is a fluidfilled
space called the matrix. The matrix connects the
space between the two membranes of the nuclear envelope,
the cell membrane, and various cytoplasmic
organelles. It functions as a tubular communication system
for transporting various substances from one part of
the cell to another. A large surface area and multiple
enzyme systems attached to the ER membranes also provide
the machinery for a major share of the cell’s metabolic
functions.
Two forms of ER exist in cells: rough and smooth.
Rough ER is studded with ribosomes attached to specific
binding sites on the membrane. The ribosomes,
with the accompanying strand of mRNA, synthesize
proteins. Proteins produced by the rough ER are usually
destined to be incorporated into cell membranes, used
in the generation of lysosomal enzymes, or exported
from the cell. The rough ER segregates these proteins
from other components of the cytoplasm and modifies
their structure for a specific function. For example, the
production of plasma proteins by liver cells takes place
in the rough ER.
The smooth ER is free of ribosomes and is continuous
with the rough ER. It does not participate in protein synthesis;
instead, its enzymes are involved in the synthesis
of lipid molecules, regulation of intracellular calcium,
and metabolism and detoxification of certain hormones
and drugs. It is the site of lipid, lipoprotein, and steroid
hormone synthesis. The sarcoplasmic reticulum of skeletal
and cardiac muscle cells is a form of smooth ER. Calcium
ions needed for muscle contraction are stored and
released from cisternae of the sarcoplasmic reticulum.
The smooth ER of the liver is involved in glycogen storage
and metabolism of lipid-soluble drugs.
Golgi Apparatus(definition and it functions): The Golgi apparatus, sometimes
called the Golgi complex, consists of stacks of thin, flattened
vesicles or sacs (see Fig. 1-4). These Golgi bodies
are found near the nucleus and function in association
with the ER. Substances produced in the ER are carried
to the Golgi complex in small, membrane-covered transport
vesicles. Many cells synthesize proteins that are
larger than the active product. The Golgi complex modifies
these substances and packages them into secretory
granules or vesicles. Insulin, for example, is synthesized
as a large, inactive proinsulin molecule that is cut apart
to produce a smaller, active insulin molecule within the
Golgi complex of the beta cells of the pancreas. In addition
to producing secretory granules, the Golgi complex
is thought to produce large carbohydrate molecules that
combine with proteins produced by the rough ER to form glycoproteins.

Figure 1-4. Three-dimensional view of the rough and the
smooth endoplasmic reticula (ER) and the Golgi apparatus. The
ER functions as a tubular communication system through
which substances can be transported from one part of the cell
to another and as the site of protein (rough ER), carbohydrate,
and lipid (smooth ER) synthesis. Most of the proteins synthesized
by the rough ER are sealed into transfer vesicles and
transported to the Golgi apparatus, where they are modified
and packaged into secretory granules.
Lysosomes(definition and it functions):
The lysosomes, which can be viewed as the digestive
organelles of the cell, are small, membrane-enclosed sacs
filled with hydrolytic enzymes. These enzymes can break
down excess and worn-out cell parts as well as foreign
substances that are taken into the cell. All of the lysosomal
enzymes are acid hydrolases, which means that they
require an acid environment. The lysosomes provide this
environment by maintaining a pH of approximately 5.0
in their interior. The pH of the cytosol and other cellular
components is approximately 7.2 and are therefore protected
from escaped lysosomal enzymes. Like all other
cellular organelles, lysosomes not only contain a unique
collection of enzymes, but also have a unique surrounding
membrane that prevents the release of its digestive
enzymes into the cytosol.
Lysosomes are formed from digestive vesicles called
endosomes. These vesicles fuse to form multivesicular
bodies called early endosomes (Fig. 1-5). The early endosomes
mature into late endosomes as they recycle lipids,
proteins, and other membrane components back to the
plasma membrane in vesicles called recycling vesicles.
Lysosomal enzymes are synthesized in the rough ER and
then transported to the Golgi apparatus, where they are
biochemically modified and packaged for transport to
the endosomes. The late endosomes mature into lysosomes
as they progressively accumulate newly synthesized
acid hydrolases from the Golgi apparatus.
Depending on the nature of the substance, different
pathways are used for lysosomal degradation of unwanted
materials. Small extracellular particles such as extracellular
proteins and plasma membrane proteins form endocytotic
vesicles after being internalized by endocytosis and
receptor-mediated endocytosis (Fig. 1-5A). These vesicles
are converted into early and late endosomes, after which
they mature into lysosomes as they accumulate newly synthesized hydrolases and vesicular proton pumps.
Large extracellular particles such as bacteria, cell debris,
and other foreign particles are engulfed in a process
called phagocytosis (Fig. 1-5B). A phagosome, formed as
the material is internalized within the cell, fuses with a
lysosome to form a phagolysosome. Intracellular particles,
such as entire organelles, cytoplasmic proteins, and
other cellular components, are engulfed in a process
called autophagy (Fig. 1-5C). These particles are isolated
from the cytoplasmic matrix by endoplasmic reticulum
membranes to form an autophagosome, which then fuses
with a lysosome to form an autophagolysosome.
Although the lysosomal enzymes can break down
most proteins, carbohydrates, and lipids to their basic constituents, some materials remain undigested. These
undigested materials may remain in the cytoplasm as
residual bodies or be extruded from the cell. In some
long-lived cells, such as neurons and heart muscle cells,
large quantities of residual bodies accumulate as lipofuscin
granules or age pigments. Other indigestible pigments,
such as inhaled carbon particles and tattoo
pigments, also accumulate and may persist in residual
bodies for decades.
Lysosomes are also repositories where cells accumulate
abnormal substances that cannot be completely
digested or broken down. In some inherited diseases
known as lysosomal storage diseases, a specific lysosomal
enzyme is absent or inactive, in which case the digestion
of certain cellular substances (e.g., glucocerebrosides,
gangliosides, sphingomyelin) does not occur. As a result,
these substances accumulate in the cell. In Tay-Sachs disease
(see Chapter 6), an autosomal recessive disorder,
hexosaminidase A, which is the lysosomal enzyme
needed for degrading the GM2 ganglioside found in nerve
cell membranes, is absent. Although the GM2 ganglioside
accumulates in many tissues, such as the heart, liver,
and spleen, its accumulation in the nervous system and
retina of the eye causes the most damage.
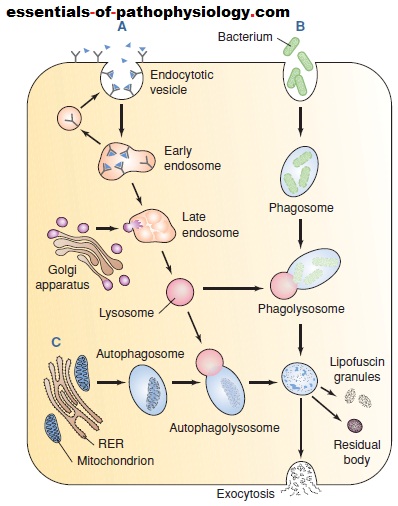
(A) Receptor-mediated endocytosis with formation of
lysosomes from early and late endosomes. Vesicle contents are
sorted in the early endosome with receptors and lipids being
sent back to the membrane. Transport vesicles carry lysosomal
enzymes to the late endosomes, converting them into lysosomes
that digest proteins and other components acquired
from the endocytotic vesicles. (B) Phagocytosis involving the
delivery of large extracellular particles such as bacteria and
cellular debris to the lysosomes via phagosomes. (C)
Autophagy is the process in which worn-out mitochondria and
other cell parts are surrounded by a membrane derived from
the rough endoplasmic reticulum (RER). The resulting
autophagosome then fuses with a lysosome to form an
autophagolysosome. Undigested material may be extruded
from the cell or remain in the cytoplasm as lipofuscin granules
or membrane-bound residual bodies.
Peroxisomes (definition and it functions):
Smaller than lysosomes, spherical membrane-bound
organelles called peroxisomes contain a special enzyme
that degrades peroxides (e.g., hydrogen peroxide). Peroxisomes
function in the control of free radicals (see Chapter
2). Unless degraded, these highly unstable chemical
compounds would damage other cytoplasmic molecules.
Peroxisomes also contain the enzymes needed for breaking
down very–long-chain fatty acids, which are ineffectively
degraded by mitochondrial enzymes. In liver cells,
peroxisomal enzymes are involved in the formation of the
bile acids.
Proteasomes (definition and it functions):
Proteasomes are small compartmentalized protein complexes
that are responsible for proteolysis of malformed
and misfolded proteins. The process of cytosolic proteolysis
is carefully controlled by the cell and requires that
the protein be targeted for degradation. This process
involves ubiquitination, a process whereby several small
ubiquitin molecules (small 76-amino-acid polypeptide
chain) are attached to an amino acid residue of the targeted
protein. Once a protein is so tagged, it is degraded
by proteasomes. After the targeted protein has been
degraded, the resultant amino acids join the intracellular
pool of free amino acids and the ubiquitin molecules are
released and recycled.
Mitochondria (definition and it functions):
The mitochondria are literally the “power plants” of the
cell because they transform organic compounds into
energy that is easily accessible to the cell. They do not
make energy, but extract it from organic compounds.
Mitochondria contain the enzymes needed for capturing most of the energy in foodstuffs and converting it into
cellular energy. This multistep process requires oxygen
and is often referred to as aerobic metabolism. Much of
this energy is stored in the high-energy phosphate bonds
of compounds such as adenosine triphosphate (ATP) that
power the various cellular activities. Mitochondria are
found close to the site of energy consumption in the cell
(e.g., near the myofibrils in muscle cells). The number of
mitochondria in a given cell type is largely determined by
the type of activity the cell performs and how much
energy is needed to undertake the activity. For example,
a dramatic increase in mitochondria occurs in skeletal
muscle repeatedly stimulated to contract.
The mitochondria are composed of two membranes:
an outer membrane that encloses the periphery of the
mitochondrion and an inner membrane that forms
shelflike projections, called cristae (Fig. 1-6). The narrow
space between the outer and inner membranes is
called the intermembrane space, whereas the large space
enclosed by the inner membrane is termed the matrix
space. The outer mitochondrial membrane contains a
large number of transmembrane porins, through which
water-soluble molecules may pass. The inner membrane
contains the respiratory chain enzymes and transport
proteins needed for the synthesis of ATP. In certain
regions, the outer and inner membranes contact each
other; these contact points serve as pathways for proteins
and small molecules to enter and leave the matrix
space.
Aucun commentaire:
Enregistrer un commentaire